徐匯區INDeCTD推薦
審評效率與時間線優化 eCTD的標準化縮短了審評周期:集中程序平均審評時間從18個月降至12個月,互認程序可在90天內完成成員國意見協調。自動化驗證工具減少了格式錯誤導致的退審率,但復雜藥學數據的科學審評仍需較長時間。申請人可通過預提交會議(Pre-submission meeting)提前溝通技術細節,規避潛在延誤。 區域協作與全球互認 歐盟通過互認程序與澳大利亞、加拿大等國實現eCTD數據共享,CEP證書在40余個非歐盟國家有效。然而,模塊一區域信息的差異性仍要求申請人定制化調整,例如亞洲國家可能要求附加穩定性研究數據。ICH的協調作用有助于減少重復提交,但完全全球化仍需解決法規和技術壁壘。 技術工具與行業生態 主流eCTD編輯軟件(如Lorenz、Extedo)支持歐盟區域模板的自動化生成,并與驗證工具集成實現一鍵校驗。云平臺解決方案逐漸普及,支持多國團隊協同編輯和實時版本控制。然而,軟件采購和維護成本較高,中小企業常選擇外包給專業服務商完成遞交。澳大利亞ANDA注冊申報相關技術支持。徐匯區INDeCTD推薦

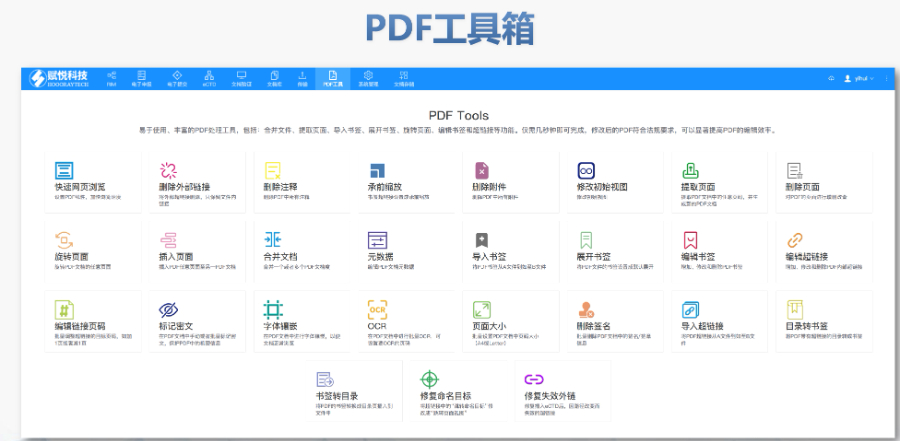
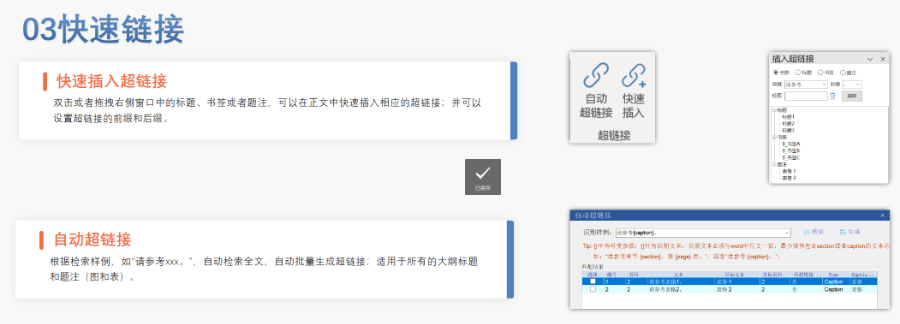
PDF工具箱 ?批量處理與格式修復 支持PDF合并、拆分、提取頁面、旋轉頁面等操作,可批量修復字體未嵌入、超鏈接錯誤等問題,確保文件符合藥品注冊法規要求。 ?智能書簽與超鏈接管理 提供書簽導入/導出、超鏈接自動生成(支持關鍵字搜索定位鏈接)、題注超鏈接拖拽式編輯等功能,簡化復雜文檔的導航設計。 ?文檔轉換與OCR識別 支持Word轉PDF(自動生成書簽、嵌入字體),以及PDF與Word、Excel等格式互轉,集成OCR功能用于掃描件文字識別。 ?合規性驗證 自動驗證PDF的頁面布局、頁碼連續性、空白頁、目錄層級等屬性,并定位具體錯誤位置,減少人工檢查成本。 ?安全與協作功能 支持文檔加密、數字簽名、云端同步及多設備共享,滿足企業級文件安全管理需求。工業園區國際注冊eCTD發布系統瑞士ANDA注冊申報相關技術支持。
生命周期管理與變更遞交 eCTD支持全生命周期管理,申請人需通過序列更(Sequence)反映藥品變更信息。例如CEP證書的更需提交“變更說明表”,對比已批準和擬修改內容,并附修訂版技術文檔。重大變更(如生產工藝調整)可能觸發GMP現場檢查,EDQM將根據風險評估決定是否啟動核查。 電子簽名與法律效力 歐盟接受符合《電子簽名法》的數字簽名,手寫簽名的掃描件需嵌入PDF并加密保護。模塊1的申請表和承諾書必須包含有效簽名,且XML文件需通過MD5校驗確保完整性。若使用第三方簽署工具,需提前向監管機構報備并獲取技術認可。 中小企業支持與資源獲取 EMA和EDQM為中小企業提供eCTD實施指南、驗證工具及培訓研討會。例如,EDQM官網發布CEP遞交模板和案例庫,EMA則定期更Q&A文檔以解答常見問題。此外,歐盟設立專項基金,資助中小企業完成eCTD轉換和系統部署。
美國電子提交通道ESG(Electronic Submissions Gateway)是美國食品藥品監督管理局(FDA)建立的電子化監管信息提交系統,旨在為制藥、生物制品、醫療器械等行業提供安全、高效的電子申報服務。自2006年啟用以來,ESG已成為FDA接收電子監管材料的入口,每日處理上千份提交文件,涵蓋上市前審批、上市后監管、臨床試驗數據、不良反應報告等多種類型。該系統通過數字證書加密和公鑰基礎設施(PKI)技術,確保文件傳輸的真實性、完整性和不可否認性,符合FDA對電子提交的嚴格合規要求。在技術層面,ESG具備強大的文件處理能力。2018年系統升級后,取消了單個文件8GB的限制,可支持高達35GB的大型文件提交,進一步滿足復雜申報需求。此外,文件格式需遵循eCTD(電子通用技術文檔)規范,包括模塊化結構、PDF標準化和XML元數據整合,以確保全球監管機構兼容性。2025年3月28日起,FDA將啟用新一代平臺ESG NextGen,逐步替代現有系統,過渡期需關注兼容性和穩定性問題。澳大利亞的eCTD申報相關技術支持。
電子簽章與傳輸安全 文件需經AES-256加密后刻錄至不可擦寫光盤,并附MD5校驗碼。光盤損壞或病毒污染將觸發重遞交流程,原載體按銷毀程序處理。 ?審評與核查協同 自2018年起,FDA要求提交兩套光盤分別用于審評和現場核查,2022年調整為“1套審評+1套核查+1套專項資料”模式,提升流程效率。 ?國際化兼容性增強 美國eCTD系統支持與歐盟、日本等地區的XML互操作性,但區域差異(如模塊1的標簽要求)仍需人工適配。 ?未來通道創 FDA計劃引入API接口支持企業系統直連,并探索基于云存儲的實時提交與審評,減少物理媒介依賴。加拿大IND注冊申報相關技術支持。南京國際注冊eCTD軟件
美國eCTD注冊外包相關技術支持。徐匯區INDeCTD推薦
仿制藥作為提高藥物可及性與可負擔性的一類藥物,2012年以前,注冊審評是不收取任何費用的,但當時仿制藥申請積壓嚴重,從申報到獲批需要3~5年的時間。 美國國會于2012年頒布了仿制藥使用者費用修正案(Generic Drug User Fee Amendments, GDUFA),該法律要求制藥行業支付一定的用戶費用,以補充仿制藥申請的審評以及現場檢查的費用,減少仿制藥申請積壓,縮短審評時間,增加基于風險的現場檢查等,其目的是加快公眾獲得安全有效的仿制藥,并降低行業成本。 GDUFA必須每五年重授權一次,于2017年更(GDUFA II),于2022年更(GDUFA III); 目前收費種類分為以下四種:ANDA審評費、DMF審評費,在審評時一次性繳納;項目費(Program fee)、設施費(Facility fee),是上市后每年繳納一次。徐匯區INDeCTD推薦
- 浦東新區原料藥eCTD文件如何制作 2025-04-26
- 南京新藥eCTD找哪家 2025-04-26
- 安徽INDeCTD找哪家 2025-04-26
- 蕪湖CDE eCTD服務電話 2025-04-25
- 杭州CDE eCTD品牌 2025-04-25
- 海南eCTD發布軟件 2025-04-25
- 閔行區賦悅科技eCTD軟件 2025-04-25
- 高新區生物制品eCTD服務電話 2025-04-25
- 太倉新藥eCTD發布系統 2025-04-25
- 化學藥品eCTD銷售電話 2025-04-25
- 黃浦區電商平臺代運營圖片 2025-04-29
- 敏感度分析軟件GOPT自動化工具包 2025-04-29
- 光明區USB攝像頭模組工廠 2025-04-29
- 樂山VOI云桌面系統產品介紹 2025-04-29
- HPE DL380 6244高頻超頻服務器供應商家 2025-04-29
- 保定手動矩陣柔性夾具配件 2025-04-29
- 呼圖壁科技營銷云數據分析與洞察平臺 2025-04-29
- 貴陽活動營銷SaaS成本 2025-04-29
- 天河區USB攝像頭模組供應商 2025-04-29
- 自動駕駛激光雷達渠道 2025-04-29